




Interviews
-


Prof. Dr. Hartmut Göbel
im InterviewMigräne: kohlenhydratreiches Frühstück ist wichtigBei Migränepatienten benötigt das Nervensystem eine hohe stetige Energieversorgung. Wichtig ist deshalb ein ausgiebiges …
-

Prof. Dr. Markus Walther
im InterviewKünstliches Sprunggelenk ist besser als VersteifungDie neuste Generation der Sprunggelenk-Endoprothesen führt zu besseren Resultaten als die Sprunggelenkversteifung. Das war nicht immer der …
-


Prof. Dr. Kirsten Müller-Vahl
im InterviewTourette-Syndrom: Tics verändern sich im Laufe der ErkrankungTics sind sehr individuell und verändern sich im Laufe des Lebens. Einige Tics sind häufiger als andere. Die ersten Tics treten schon bei …
-


Prof. Dr. Claudia Trenkwalder
im InterviewSchlafstörungen sind typisch bei Restless-Legs-SyndromSchlafstörungen oder auch Durchschlafstörungen sind typisch für RLS und treten meist zu bestimmten Tageszeiten auf. …
-


Prof. Dr. Bernd Böttiger
im InterviewPlötzlicher Herztod ist dritthäufigste TodesursacheMehr als 70.000 Menschen sterben in Deutschland jedes Jahr an Herzversagen. Die Überlebensrate bei einem plötzlichen Herztod liegt bei nur …
-

Prof. Dr. Georgi Wassilew
im InterviewHüftdysplasie kann vorzeitig zu Arthrose führenBleibt eine Hüftdysplasie unbehandelt, kann sie langfristig zu einer Abnutzung des Hüftgelenks und somit zu vorzeitiger Arthrose führen. …
















Dann empfehlen Sie Ihn weiter.
mit Patienten und Experten.





Fragen und Antworten aus dem Forum
sagt Sabine1105 vor 3 Tage zum Thema HNO (Hals-Nasen-Ohren) 1004
sagt Dr. Alamouti vor 4 Tage zum Thema Haut - Dermatologie 60
Wenn ihr Körper gegen eine Entzündung kämpft,
können diese LK anschwellen.
Bitte nicht ... mehr
fragt manilani2006 vor 4 Tage zum Thema Urologie 8
ich habe seit Wochen auf meiner Vorhaut ein pickelartiges brauner Punkt, was keinerlei Beschwerden macht. Es juckt nicht, es brennt ... mehr
sagt IcySpicy99 vor 5 Tage zum Thema Allgemeinchirurgie 273634
Ich bekam im November 2021 eine Analfissur. Diese ... mehr
fragt MAST8405 vor 6 Tage zum Thema MKG (Mund-Kiefer-Gesicht) 18
fragt Harry1955 vor 6 Tage zum Thema Orthopädie 28
fragt Tamara91 vor 7 Tage zum Thema HNO (Hals-Nasen-Ohren) 25






Häufige Symptome
-
 Abgeschlagenheit
AbgeschlagenheitAbgeschlagenheit beschreibt einen Zustand, in dem sich eine Person allgemein geschwächt, energielos, erschöpft und müde fühlt. Das …
-
 Augenzucken: Häufige Ursachen
Augenzucken: Häufige UrsachenUnter Augenzucken versteht man unkontrollierte Bewegungen oder Krämpfe des Augenlids oder der Augenmuskeln. Es gibt verschiedene Arten von …
-
 Blähungen
BlähungenBlähungen sind stark vermehrte Ansammlungen von Gas im Darm. Der Fachbegriff für zu viel Luft im Bauch und häufigen Windabgang lautet …
-
 Blut im Stuhl
Blut im StuhlUnter dem Begriff Blut im Stuhl versteht man sichtbare oder unsichtbare Beimengungen oder Auflagerungen von Blut im Stuhl (Kot, Fäzes). Je …
-
 Haarausfall
HaarausfallUnter dem Begriff Haarausfall (Effluvium) versteht man einen übermäßigen Verlust an Haaren, der zur Glatzenbildung (Haarlosigkeit, …
-
 Muskelkrämpfe
MuskelkrämpfeMuskelkrämpfe können den Alltag erheblich einschränken. Bei jungen Erwachsenen, insbesondere bei Sportlern, treten sie gelegentlich auf. …
-
 Nervosität (Unruhe)
Nervosität (Unruhe)Nervosität ist ein Zustand der inneren Unruhe und Anspannung. Vielfach ist Nervosität der Situation geschuldet und lässt sich leicht …
-
 Vergesslichkeit
VergesslichkeitDas Gedächtnis ist ein extrem komplexes System unseres Gehirns, das bis heute noch nicht ins Letzte verstanden ist. Das Kurzzeitgedächtnis …








Häufige Krankheiten
-
 Asthma bronchiale
Asthma bronchialeAsthma bronchiale oder Bronchialasthma bedeutet eine zeitweise krankhafte Enge der Bronchien. Dabei kommt es zu einer typischen …
-
 Bluthochdruck
BluthochdruckDer Bluthochdruck (Hypertonie, arterielle Hypertonie) ist eine sehr häufige Störung und gehört zu den Zivilisationskrankheiten. Ein zu …
-
 Gastritis (Magenschleimhautentzündung)
Gastritis (Magenschleimhautentzündung)Eine Magenschleimhautentzündung, die in Fachkreisen auch als Gastritis, Gastroenteritis (wenn der Darm beteiligt ist) sowie …
-
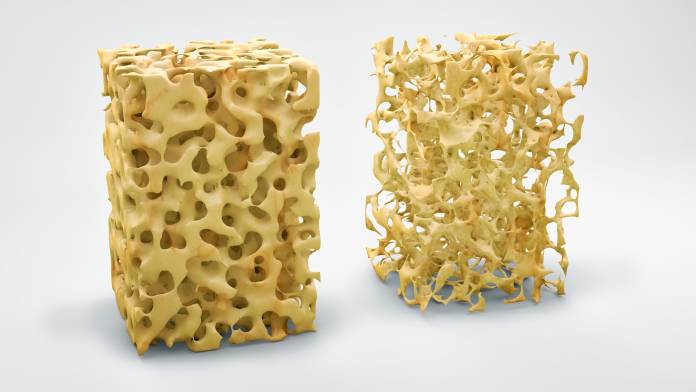 Osteoporose
OsteoporoseDie Osteoporose ist eine Erkrankung, bei der es wegen einer Abnahme der Knochensubstanz zur verminderten Stabilität der Knochen kommt. Sie …
-
 Rheuma (Rheumatoide Arthritis)
Rheuma (Rheumatoide Arthritis)Die rheumatoide Arthritis (RA) oder chronische Polyarthritis (CP, auch: primär chronische Polyarthritis) ist eine Erkrankung, bei der es zu …
-
 Schilddrüsenunterfunktion
SchilddrüsenunterfunktionEine Schilddrüsenunterfunktion (Hypothyreose) begründet sich auf eine zu geringe Wirkung der Schilddrüsenhormone. In den allermeisten …






Fachgebiete
- Augenheilkunde
- Gynäkologie (Frauengesundheit)
- Hals-Nasen-Ohren Heilkunde
- Dermatologie (Hautgesundheit)
- Kardiologie (Herzgesundheit)
- Infektionen
- Innere Medizin
- Krebs (Onkologie)
- Mund-Kiefer-Gesichtschirurgie
- Neurologie
- Orthopädie
- Psychiatrie
- Radiologie
- Urologie
- Zahnmedizin
Ratgeber Operation
-
 Ratgeber Operation
Ratgeber OperationSie müssen operiert werden? Lesen Sie unseren Ratgeber und erfahren Sie, was Sie vor und nach einer Operation beachten sollten. …
-
 Packliste Krankenhaus
Packliste KrankenhausEs gibt eine Reihe Dinge, an die man bei einem geplanten Krankenhausaufenthalt denken muss. Mit einer Packliste für das Krankenhaus ist es …
-
 Ablauf einer Operation
Ablauf einer OperationDer Ablauf einer Operation gestaltet sich sehr verschieden, je nachdem, welcher Eingriff vorgenommen wird. In einigen Grundzügen sind sich …
-
 Nach der Operation
Nach der OperationIst eine Operation überstanden, sind meist weitere Therapien und Untersuchungen notwendig, um eine vollständige Genesung zu …




Chirurgie
- Ratgeber Operation
- Allgemeinchirurgie
- Gefäßchirurgie, Venenchirurgie
- Handchirurgie
- Herzchirurgie
- Intensivmedizin
- Kinderchirurgie
- Neurochirurgie
- Plastische Chirurgie (Schönheitschirurgie)
- Thoraxchirurgie
Orthopädische Erkrankungen
-
 Arthrose
ArthroseArthrose bedeutet übersetzt Gelenkerkrankung. In der Medizin wird als Arthrose ein zunehmender, auch altersabhängiger Knorpelabrieb der …
-
 Rückenschmerzen
RückenschmerzenRückenschmerzen sind Beschwerden, die in der Bevölkerung sehr weit verbreitet sind. Über 80 Prozent der Deutschen leiden in ihrem Leben …
-
 Bandscheibenvorfall
BandscheibenvorfallBei einem Bandscheibenvorfall (Bandscheibenprolaps, Diskusprolaps) tritt ein Teil der elastischen Bandscheibe, die sich als mechanischer …
-
 Karpaltunnelsyndrom
KarpaltunnelsyndromDas Karpaltunnelsyndrom bezeichnet eine Verengung einer tunnelförmigen Struktur zwischen Knochen und Bändern an der Beugeseite des …
-
 Fersensporn
FersenspornDer Fersensporn ist eine Erkrankung mit dornartigen knöchernen Ausziehungen am Fersenbein. Es handelt sich um eine schmerzhafte Erkrankung, …
-
 Meniskusverletzung
MeniskusverletzungDer Meniskus ist ein knorpeliges Gebilde im Gelenk. Jedes Knie hat einen Außenmeniskus und einen Innenmeniskus. Die Menisken haben die Form …
-
 Kreuzbandriss
KreuzbandrissBei starker mechanischer Belastung kann das vordere Kreuzband oder das hintere Kreuzband innerhalb des Knies reißen (Kreuzbandruptur). Es …
-
 Zehenbruch
ZehenbruchWas ist ein Zehenbruch? Der Zehenbruch (Zehenfraktur) gehört zu den häufigeren Knochenbrüchen. Gebrochene Zehen können die Folge einer …








Blutwerte leicht und verständlich erklärt
-
 Was ist ein Blutbild?
Was ist ein Blutbild?Wer sich ein Bild über das Blut machen will, der braucht ein Blutbild. Diese Aussage ist zwar nicht falsch, aber auch nicht …
-
 Kleines Blutbild - einfach erklärt
Kleines Blutbild - einfach erklärtZu den häufigsten Untersuchungen in der Medizin gehört das kleine Blutbild. Das kleine Blutbild liefert dem Arzt einen Überblick über …
-
 Großes Blutbild
Großes BlutbildDas große Blutbild ist wie auch das kleine Blutbild eine Standarduntersuchung in der Medizin. Viele Patienten denken, dass beim großen …
-
Rote Blutkörperchen (Erythrozyten)
Die Hauptaufgabe der roten Blutkörperchen ist der Transport von Sauerstoff. Auf der einen Seite sorgen sie dafür, dass der Sauerstoff von …
-
Weiße Blutkörperchen (Leukozyten)
Weiße Blutkörperchen sind für die Immunabwehr und die Abwehr von Krankheitserregern zuständig. Es gibt verschiedene Arten von weißen …
-
Hämoglobin
Hämoglobin (Hb) ist das wichtigste Eiweiß der roten Blutkörperchen (Erythrozyten). Hämoglobin enthält Eisen und ist verantwortlich für …



Schwangerschaft
-
 Kalender über die Schwangerschaft
Kalender über die SchwangerschaftEine Schwangerschaft ist aufregend, aber auch aufwühlend zugleich. Natürlich überwiegt zunächst die Freude, wenn die ersten Anzeichen …
-
 Beschwerden in der Schwangerschaft
Beschwerden in der SchwangerschaftÜbelkeit und Erbrechen kennt beinahe jede Schwangere. Doch gehören diese zu den harmloseren Beschwerden während der Schwangerschaft. …
-
 Untersuchungen in der Schwangerschaft
Untersuchungen in der SchwangerschaftWährend der Schwangerschaft werden verschiedene Untersuchungen durchgeführt. Neben Ultraschall können auch andere Methoden sinnvoll …
-
 Natürliche Geburt
Natürliche GeburtDie natürliche Geburt ist die Geburtsmethode, das Kind über den herkömmlichen Weg durch den Gebärmutterhals und die Scheide zu bekommen. …




News

Viele der bisher eingesetzten Antibiotika verlieren ihre Wirkung. Bakterien werden gegen sie resistent. Das bedeutet, dass diese Antibiotika bakterielle Infektionen nicht mehr bekämpfen können. Schätzungen zufolge sterben in der EU jedes Jahr 33 …

GLP-1-Agonisten wie Semaglutid und Tirzepatid sind Medikamente zur Behandlung von Übergewicht. Sie haben sich als wirksam für eine starke Gewichtsabnahme erwiesen. Sie wurden urprünglich zur Behandlung von Diabetes eingesetzt. Studien deuten …

Die von der STIKO empfohlene neue Impfung Qdenga® könnte ein Fortschritt in der Bekämpfung der Erkrankung sein. Diese Empfehlung ist von …

23.08.23 - Die Affenpocken, inzwischen in Mpox umbenannt, breiten sich in Europa wieder aus. Nach einem Anstieg der Infektionen im Frühjahr 2022 wurden dem RKI etwa 3.700 Fälle übermittelt. Von Anfang 2023 bis Juli 2023 ebbten die Fallzahlen …

18.08.23 - Das humane Papillomavirus (HPV) ist ein weit verbreitetes Virus. Es wird durch Haut-zu-Haut-Kontakt übertragen. Eine neue Metastudie hat gezeigt, dass weltweit fast jeder dritte Mann mit HPV infiziert ist. Da einige dieser Viren Krebs …
Plastische Chirurgie

Die Brustvergrößerung gehört zu den beliebtesten Schönheitsoperationen. Aber auch eine Schönheitsoperation ist nicht ohne Risiko. Erfahren Sie mehr. …

Die Nasenkorrektur gehört zu den kompliziertesten Eingriffen der Plastischen Chirurgie. Wer sich dafür entscheidet, sollte einen Spezialisten aufsuchen. …

Eine Hautstraffung beziehungsweise Faltenstraffung im Bereich des mittleren und unteren Gesichtes oder des Halses nennt man unteres Facelift. Wenn man von einem Facelift spricht, dann meint man damit meistens das untere Facelift. Es bestehen bei …

Reiterhosen? Bauch trotz Training? Die Fettabsaugung dient zur Korrektur von Körperkonturen. Abnehmen kann man damit allerdings nicht. …

Straffung der Oberschenkel, Straffung des Gesäßes
Wenn im Gesäß- und Oberschenkelbereich durch überschüssige Haut und Unterhautfett eine Erschlaffung bemerkbar ist, lässt sich diese operativ straffen. Je nach Befund kann die Operation am Po, an der Oberschenkelinnen- sowie Außenseite oder in …
Häufige Fragen
- Achillessehnenriss: Was muss man nach der Operation beachten?
- Inwiefern kann Zucker Nierensteine verursachen?
- Gibt es einen Zusammenhang zwischen Keratokonus und ADHS?
- Wie funktioniert der Perthes-Test?
- Woran erkennt man einen Arterienverschluss in Beinen und Armen?
- Wie lange ist man nach einem Kreuzbandriss krankgeschrieben?
- Welche Hautveränderungen treten bei Diabetes häufig auf?
Häufige Operationen
- Steißbeinfistel (Sinus pilonidalis)
- Leisten- und Schenkelbruch
- Blinddarmentzündung
- Hämorrhoiden Behandlung
- LASIK (Augen lasern statt Brille)
- Bauchspiegelung
- Gebärmutterentfernung
- Stentprothese
- Kniegelenk-Prothese
Themen im Fokus
-
 Alternative Medizin
Alternative MedizinDie Naturheilkunde wird alternativ oder ergänzend zur Schulmedizin eingesetzt. Immer mehr Menschen schwören auf die sanfte Behandlung …
-
 Blutwerte
BlutwerteBlutwerte wie Cholesterin, Blutzucker, Eisen-, Leber- und Nierenwerte, sowie Schilddrüsen- und Entzündungswerte sind entscheidend für die …
-
 Reisemedizin
ReisemedizinWer gerne auf Reisen ist, muss sich vor Infektionen schützen. Viele Infektion kommen bei uns nicht vor, treten aber in fremden Ländern …



Letzte Aktualisierung am 03.12.2009.